Elisaveta Miladinova
Email: e_miladinova@mail.bg
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 4 ISSUE: 2
Page No: 353-364
Elisaveta Miladinova
Email: e_miladinova@mail.bg
Elisaveta Miladinovaa, b*, Peicho Petkova, Nevena Ilievac and Leandar Litova
a Faculty of Physics, Sofia University "St. Kliment Ohridski", 5 James Bourchier Blvd., 1164 Sofia, Bulgaria
b Section of Biosimulation and Bioinformatics, Center for Medical Statistics, Informatics and Intelligent Systems, Medical University of Vienna, Spitalgasse 23, 1090 Vienna, Austria
c Institute of Information and Communication Technologies, Bulgarian Academy of Sciences, Acad. G. Bonchev St., Block 25A, 1113 Sofia, Bulgaria
Elisaveta Miladinova, Computer aided study of the oxytocin - receptor complex binding sites(2020)Journal of Computational Chemistry & Molecular Modeling 4(2)
Oxytocin (OT) is a neurohypophysial hormone, which acts both on the peripheral tissues and as a neurotransmitter in the brain. It plays an important role in the control of uterine contractions during labor, secretion of milk and regulates many social and behavioural functions. OT accomplishes its functions via interaction with specific oxytocin receptors (OTRs), which belong to the rhodopsin type (family A) group of G-protein coupled receptors (GPCRs). Increased oxytocin secretion associated with hormonal abnormalities in endocrine glands (thyroid, pituitary) during pregnancy may cause premature birth. Oxytocin receptor antagonists are widely employed for prevention in such cases. Design of these antagonists requires a proper model of the hormone – receptor interaction. However, the 3D structure of the OTR is not known.
The aim of this investigation is to construct a three - dimensional model of the oxytocin receptor, to be used to define the binding sites between oxytocin and its receptor, as a prerequisite for further search for oxytocin antagonists. We employ homology modeling as an efficient technique to generate a family of oxytocin GPCRs structures. The best structure is investigated for its interactions with the hormone oxytocin through molecular dynamics (MD) simulations. The potential oxytocin - receptor binding sites are determined. The structural changes in the receptor induced by oxytocin binding are investigated.
Keywords: homology modelling; molecular dynamics (MD); oxytocin (OT); oxytocin binding sites; oxytocin receptor (OTR); 3D models.
Oxytocin is a hormone with a peptide structure, which acts both on the peripheral tissues (hormonal), and as a neurotransmitter in the brain. It is synthesized from neurons located in the hypothalamus [1]. Oxytocin accomplishes its functions via interaction with specific receptor, which belongs to the rhodopsin-type (class I) group of G-protein coupled receptors, by far the largest and most diverse GPCR subfamily [2, 3]. The hormone binds to the transmembrane part of the receptor and thus activates it, causing the start of various intracellular signaling pathways. This leads to an increase in the concentration of Ca²+ ions in the cell, actin filaments bind to myosin resulting in contraction of the smooth muscle of the uterus. Increased oxytocin secretion associated with some hormonal abnormalities in endocrine glands during pregnancy may cause uterus contractions and premature birth. In such cases oxytocin receptor antagonists are widely employed for prevention [4, 5], however their efficiency is limited.
As target for many drugs the G protein - coupled receptors are important class of transmembrane (TM) proteins [6, 7]. All members of the GPCRs share structural and functional similarities. They are built from single polypeptide chain, which is composed of seven membrane-spanning α-helices (TM1-7) connected by alternating extracellular (EL1, EL2 and EL3) and intracellular (IL1, IL2 and IL3) loop domains. At all GPCRs the N-terminal part is exposed to the extracellular space and C-terminal tail is located in the intracellular fluid (cytosol) of the cell. The C-terminal portion of the receptor participate in interaction with cytosolic G proteins. An essential disulfide bond, that is formed between two cysteine residues, situated in the TM3/EL1 interface and EL2, plays an important role in the packing and stabilization of the seven TM domains of GPCRs. These cysteine amino acid residues are conserved in almost all receptor structures (Figure 1). The members of the GPCRs differ mainly in the length of the amino acid sequence, their intracellular loops, as well as in length and function of their N- and C-terminal domains. These terminal parts provide specific interactions with the various receptor proteins in extracellular and intracellular environment. Despite these differences in several subfamilies is found significant sequence homology.
The GPCRs transfer extracellular signals to the intracellular targets by binding the agonistic ligands to their extracellular part. This elicits conformational changes of the receptor and activates the G protein [8, 9].
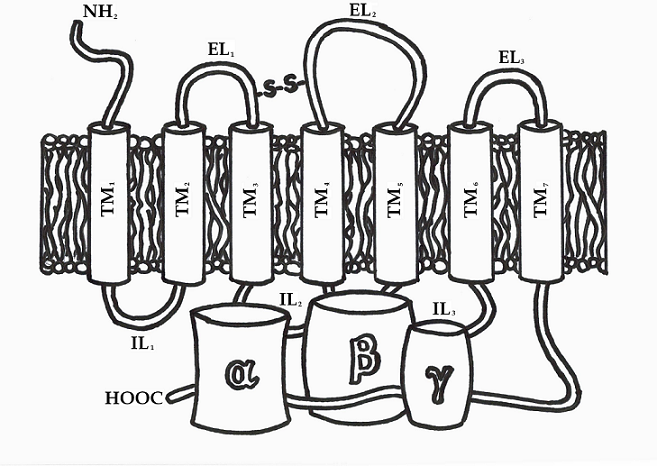
Figure 1. GPCR structure linked to a trimeric guanine-nucleotide binding protein.
There are over 800 unique GPCRs known by now. Based on the degree of sequence homology and functional similarity, these receptors are grouped into five different subfamilies: rhodopsin (family A), secretin (family B), glutamate (family C), adhesion and Frizzled/Taste2. The rhodopsin family is by far the largest and its members are characterized by conserved sequence motifs that imply shared structural features and activation mechanisms [2, 3]. Until now, there is very little structural information available about this subfamily of proteins as they are membrane proteins and thus difficult to crystallize. In order to design rationally an effective drug, which to act on any given target protein, knowledge of the three-dimensional (3D) structure of the target receptor and the binding sites with its ligand is highly desirable [10]. Unfortunately, the 3D structure of the oxytocin receptor is unknown.
The aim of the current investigation is to construct a three-dimensional model of the oxytocin receptor and to define the binding sites by studying complex formation with its natural ligand – oxytocin. The new model will allow the design and the development of more efficient and selective oxytocin-receptor antagonists.
2.1. Homology modelling
Homology or comparative modelling of proteins is a technique that allows the construction of a target protein model based on its amino acid sequence and some known 3D structure of a homologous protein – template [11, 12]. The template structure must be of a reasonable quality – with resolution better than 2.5 Å, and a sequence identity with the target protein of at least 25%. Also the biological function of the two proteins must be similar or at least related [13].
In this study the input sequence of human oxytocin receptor (FASTA format) was retrieved from the NCBI database [14]. The NCBI-Protein BLAST server [15] was used for the search of closest homologue of this target sequence, which contains 348 amino acid residues. Top-ranked template sequences, determined by BLAST, on the basis of optimized E-value of the target sequence, were subjected to multiple sequence alignment. The reciprocal position of the conserved amino acid residues and the alignment score both were used to select the best alignment among all possible ones.
In the present study, the 3D structure of bovine rhodopsin (PDB ID 1JFP [16, 17]) was used as a template. The MODELLER software suit for comparative protein structure modelling [18] was used in the model-building process. More specifically, the sequence alignment was performed with align2d function. The homology models of the human oxytocin receptor were generated with the help of automodel class. The final selection was guided by MODELLER objective function molpdf and the energy profile analysis in terms of DOPE (Discrete Optimized Protein Energy) score. The DOPE potential [19] characterizes the interactions between pairs of atoms and can be decomposed into a score per residue. The energy profile so obtained is useful to detect local regions of high pseudo-energy that usually correspond to errors in the model.
2.2. Membrane bilayer model
Oxytocin receptor is a transmembrane protein, thus for studying its dynamics it should be considered in complex with the membrane. A membrane bilayer model (consisting of 5 different types of phospholipids) was built for this purpose with the aid of the Membrane Builder tool within the CHARMM GUI web-based graphical user interface [20-22].
2.3. Molecular dynamics simulations
The energy minimization (EM) of the constructed oxytocin receptor structure was performed aiming at structure refinement and stability. The receptor-membrane complex was studied via MD simulations. The simulations were performed with GROMACS 5.0.7 package [23], with CHARMM 36 force field [24]. In all MD simulations a leap-frog integrator was employed. All bonds were constrained by LINCS algorithm. The cutoff radius of Coulomb interactions was set at 12 Å. The protocol comprised initial energy minimization (steepest descent algorithm), followed by equilibration for 375 ps and a production run of 300 ns, with a time-step of 2 fs. The TIP3P water model of solvent was employed. The pressure and temperature were fixed at 1 bar and 310 K by means of Parrinello-Rahman barostat and v-rescale thermostat, respectively. Periodic boundary conditions were applied in all directions. The van der Waals interactions were smoothly switched off at 10-12 Å by a force-switching function [25]. The long-range electrostatic interactions were calculated using the particle-mesh Ewald [26] method. The receptor- membrane system was then subjected to MD simulation (300 ns) with the same settings. The aim of this first simulation was to relax the membrane and solvent residues that form the interface between the receptor model and the rest of the bilayer-solvent environment. Finally, MD simulations of the whole complex membrane-oxytocin receptor-oxytocin were performed. For the energy minimization, the steepest descent method in combination with conjugate gradient method was used. The equilibration simulations in NVT and NPT ensembles with duration 0.5 ns were performed, followed by a production run of 100 ns. During the NPT equilibration, the structure of the oxytocin - receptor complex was relaxed. The other parameters were the same as for the receptor-membrane simulations.
3.1. Homology modelling
The 3D structure of the human oxytocin receptor is not known. We employed homology modeling for its determination. In order to identify the amino acid residues that form the seven transmembrane α-helices of the query protein the sequence of oxytocin receptor was aligned to those of the best template (bovine rhodopsin PDB ID 1JFP). The degree of sequence similarity between the both structures (target protein and template) is important for the accuracy of the predictions through comparative modelling. In our case, the alignment score was 74.3 bits, query coverage was 83% and sequence identity 22% (obtained by using the BLAST program) [15]. Sequential alignment between the primary amino acid sequences of the target protein (oxytocin receptor) and the template (bovine rhodopsin) was also performed with the MODELLER software package (see Figure 2). The sequence identity was 27%. The members of GPCRs family shared common motifs, which are the extracellular disulfide bridge between TM3 and EL2, Asn1.50, Asp2.50, the E/DRY motif (Asp/Glu3.49, Arg3.50, and Tyr3.51), Trp4.50, the two proline residues Pro5.50 and Pro6.50, and the NPXXY motif (Asn7.49, Pro7.50, and Tyr7.53) in TM7. The highly conserved amino acid residues in these important motifs guided the alignment, because they are related to structurally or functionally defining parts of the target protein [2]. The MODELLER align2d function, which takes into account structural information from the template, was employed to perform the sequence alignment. It uses a variable gap penalty function that places gaps in solvent- exposed and curved regions, outside secondary structure segments [27]. Thus, the alignment errors are reduced by about one third compared to those that occur with standard sequence alignment techniques. Based on the alignment with sequence identity of 27%, five homology models were generated. The structure with the lowest values of the MODELLER objective function molpdf (5399) and DOPE score (-33055), as well as the smallest number of restraints violations was accepted as a prospective 3D model and employed in the further studies (Figure 3).

Figure 2. Alignment of the human oxytocin receptor (query) and bovine rhodopsin receptor (PDB 1JFP) amino acid sequences.
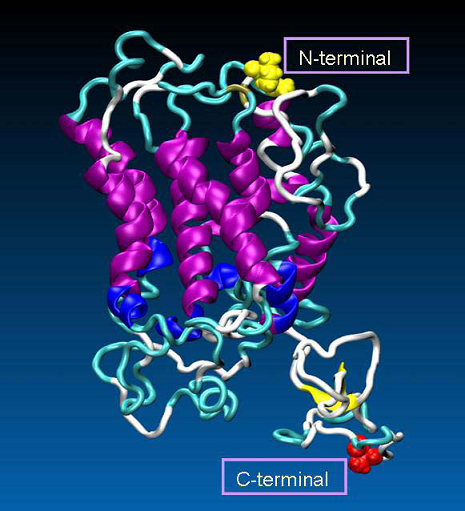
Figure 3. The 3D model of human oxytocin receptor, generated by MODELLER. The secondary structures of the receptor are colored as follows: α-helix (purple), 3-10-helix (blue), extended-β (yellow), turn (cyan), coil (white). Additionally, as VDW spheres are shown N- (in yellow) and C-terminal part (in red) of the receptor.
The geometrical and structural consistency of the constructed model were first evaluated with DOPE assessment function, as implemented in MODELLER, by comparing the energy profiles of the model and the template (Figure 4). The large peak in the template profile in the region of position 70 is most probably related to an active center. Therefore, a possible error indicated by the evaluation function, is not necessarily real.
The degree of sequence identity between target and template, however, is not the only criterion for quality assessment of the model. The choice of template with enough good resolution during homologous modeling also affects the quality of the model. A quantitative measure of the alignment is the root mean square deviation (RMSD) between the two structures,
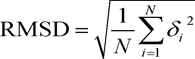
where δi is the distance between the corresponding superimposed atoms. The alignment is good if RMSD ˂ 2 Å. The template structure (bovine rhodopsin, PDB ID 1JFP) used in our study had a resolution of 1.0 Å. The model of human oxytocin receptor revealed an excellent agreement with the experimentally determined 3D structure of bovine rhodopsin, with a Cα RMSD of only 1.05 Å (Figure 5).
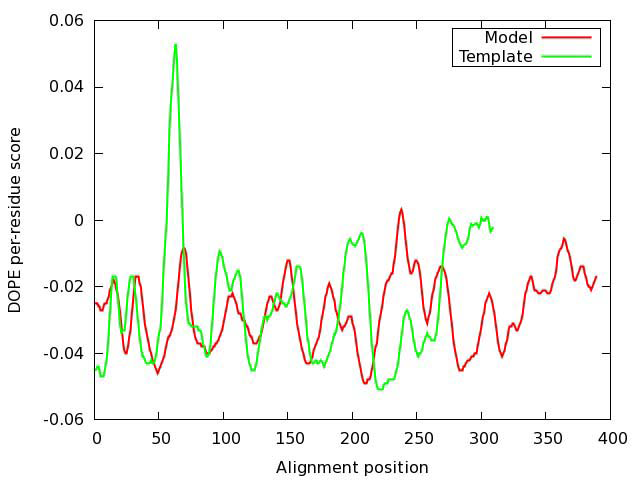
Figure 4. Evaluation of the model of oxytocin receptor. DOPE (Discrete Optimized Protein Energy) score profiles for the model and template.
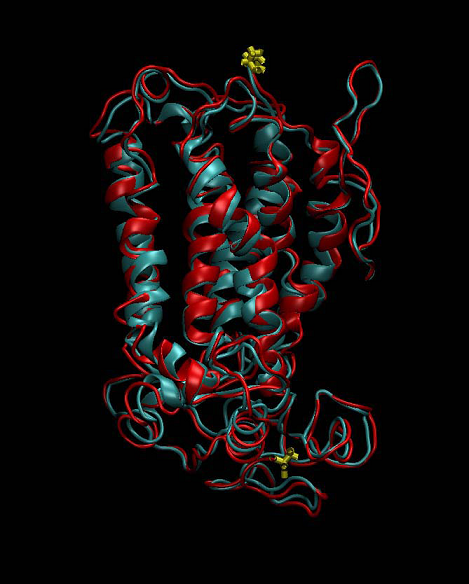
Figure 5. Superimposed structure of the template bovine rhodopsin (red) and the homology model of human oxytocin receptor (cyan). N- and C-terminal parts of both receptor structures are colored in yellow.
3.2. Membrane bilayer model
The conformational stability of GPCRs as membrane proteins strongly depends on their natural environment - the lipid bilayer [28]. For investigation of the binding mechanism of oxytocin to its receptor, a membrane model (smooth uterine muscle) was constructed, consisting of phosphatidyl choline 50.3%, phosphatidyl ethanolamine 24.8%, sphingomyelin 13.8%, phosphatidyl serine 6.4% and cholesterol 2% [29] (Figure 6).
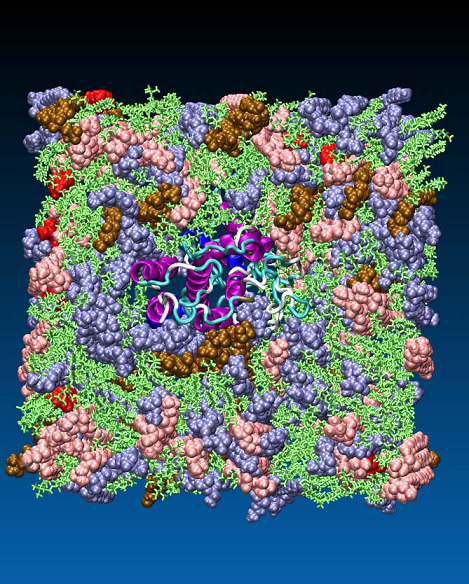
Figure 6. Model of myometrial membrane with a phospholipid composition as follows: phosphatidyl choline 50.3%, (green), phosphatidyl ethanolamine 24.8% (blue), sphingomyelin 13.8% (rose), phosphatidyl serine 6.4% (ochre) and cholesterol 2% (red).
3.3. Oxytocin - receptor complex: construction, stability and dynamics studies
The presence of doubly-charged cations, such as Zn2+ or Cu2+ play an essential role in the specific binding of OT to its receptor. However, it is unclear, whether the metal ions primarily interact with the receptor, OT, or both participants in the process [30]. There are both experimental and theoretical evidence indicating that oxytocin undergoes substantial conformational changes due to interaction with Zn2+ [31 - 33]. In the presence of Zn2+, the side chains of Ile3, Gln4, and Asn5 of OT line up to form a cohesive, near-planar surface amenable for coordination with the receptor. These changes permit binding of specific residues in OT with specific residues in its receptor. However, there is no crystallographic information available for oxytocin in complex with metal ion. Thus in order to clarify the role of the divalent ions in the oxytocin – receptor interaction we built a model of such a complex. Stability of the complex was confirmed by a series of molecular dynamics simulations [34]. In our structure the Zn2+ ion forms a near-perfect octahedral complex with six of the backbone carbonyl oxygens (associated with Tyr2, Ile3, Gln4, Cys6, Leu8 and Gly9) in the OT (Figure 7), in an agreement with the findings in [35].
Experimental data showed, that residues located in the transmembrane domains, as well as residues in extracellular domains of the receptor are involved in the binding of the peptides like OT and arginine vasopressin (AVP). The amino acid Val115 (helix 3) plays a crucial role in ligand selectivity. The highly conserved Gln residues in the transmembrane domains 3 and 6 create a common agonist-binding pocket to all different subtypes of the rhodopsin-type (class I) receptor family [1].
The oxytocin - metal ion complex is positioned by docking with VMD [36] (VsLab plug-in [37]) within the transmembrane part of the receptor taking into account its experimentally known binding sites. The OT-OTR complex is shown in Figure 8.
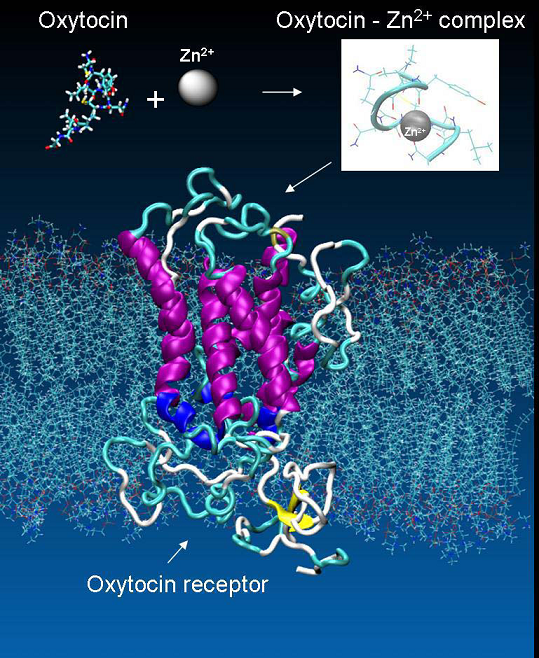
Figure 7. The metal ions as mediator for the specific binding of OT to its receptor.
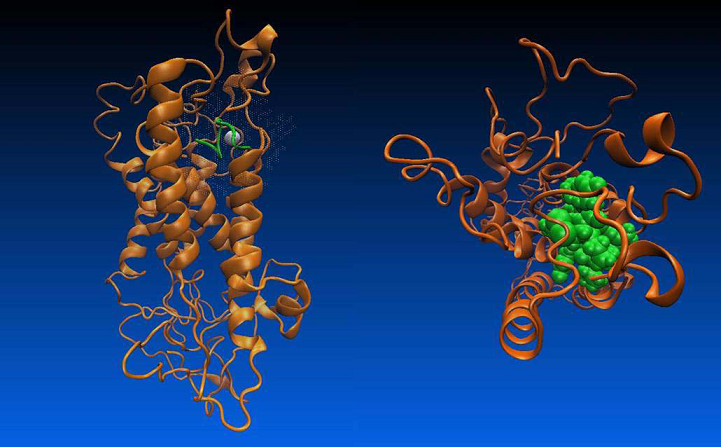
Figure 8. Oxytocin at the binding site of the oxytocin receptor - a/ side view; b/ top view.
3.4. Determination of the oxytocin binding sites
Both the hydrophobic contacts and hydrogen bonds contribute to the final score of a particular protein-ligand interaction. The tool g_mmpbsa [38], implemented in Gromacs was used to calculate binding energies of the oxytocin - OTR complex from MD trajectory. The nonpolar contacts between the receptor and the ligand were determined by decomposing the total calculated binding energy into contributions per residue by the g_mmpbsa tool. The hydrogen-bonds analysis of the complex was performed by using the computer algorithm HBonanza [39]. The information about the type of each interaction and the amino acids involved in formation of binding pocket between OTR and its ligand oxytocin is summarised in Table 1.
In total, 21 binding site residues were defined as potential interaction points in our model of OTR: Glu307 (el3), Val115 (helix 3), Tyr200 (el2), Gln119 (helix 3), Met123 (helix 3), Ile201(el2), Gln171 (helix 4), Phe291 (helix 6), Leu317 (helix 7), Val294 (helix 6), Gln295 (helix 6), Ile313 (helix 7), Val314 (helix 7), Trp300 (el3), Ile312 (helix 7), Phe311 (helix 7), Ile204 (helix 5), Trp203 (el2), Thr202 (el2), Ser298 (el3) and Trp297 (el3). The conserved residues participating in the binding of OT to its receptor are: Val115 (helix 3), Gln119 (helix 3), Met123 (helix 3), Gln171 (helix 4), Phe291 (helix 6), Leu317 (helix 7), Gln295 (helix 6). The experimental data suggests, the important role of the amino acid isoleucine in the OT for the specific binding of the oxytocin molecule to its receptor [40]. Correspondently, in our model the OTR residues binding to the Ile3 were considered crucial for the OT recognition. These residues are: Phe291 (helix 6), Leu317 (helix 7), Val294 (helix 6), Met123 (helix 3), Gln295 (helix 6) and Gln119 (helix 3). The conserved Gln residues Gln119, Gln171 and Gln295 were determined as important residues for the ligand binding and stabilization of the receptor ligand complex, which is in agreement with both theoretical and experimental results of other research groups [41, 42, 43, 44].
Table 1. Description of the oxytocin binding sites in the proposed OTR model.
|
No. |
OT |
OTR |
Intermolecular interaction |
|
1 |
Cys1 |
Glu307 (el3) |
IONIC HB |
|
2 |
Cys1 |
Val115 (helix 3) |
VDWb |
|
3 |
Cys1 |
Tyr200 (el2) |
HBa |
|
4 |
Cys1 |
Gln119 (helix 3) |
HB |
|
5 |
Tyr2 |
Met123 (helix 3) |
VDW |
|
6 |
Tyr2 |
Gln119 (helix 3) |
VDW |
|
7 |
Tyr2 |
Val115 (helix 3) |
VDW |
|
8 |
Tyr2 |
Tyr200 (el2) |
VDW |
|
9 |
Tyr2 |
Ile201(el2) |
VDW |
|
10 |
Tyr2 |
Gln171 (helix 4) |
HB |
|
11 |
Tyr2 |
Gln171 (helix 4) |
VDW |
|
12 |
Ile3 |
Phe291 (helix 6) |
VDW |
|
13 |
Ile3 |
Leu317 (helix 7) |
VDW |
|
14 |
Ile3 |
Val294 (helix 6) |
VDW |
|
15 |
Ile3 |
Met123 (helix 3) |
VDW |
|
16 |
Ile3 |
Gln295 (helix 6) |
HB |
|
17 |
Ile3 |
Gln119 (helix 3) |
HB |
|
18 |
Gln4 |
Ile313 (helix 7) |
VDW |
|
19 |
Gln4 |
Val314 (helix 7) |
VDW |
|
20 |
Gln4 |
Leu317 (helix 7) |
VDW |
|
21 |
Asn5 |
Trp300 (el3) |
VDW |
|
22 |
Asn5 |
Ile312 (helix 7) |
VDW |
|
23 |
Asn5 |
Ile313 (helix 7) |
VDW |
|
24 |
Asn5 |
Phe311 (helix 7) |
VDW |
|
25 |
Asn5 |
Ile313 (helix 7) |
HB |
|
26 |
Asn5 |
Phe311 (helix 7) |
HB |
|
27 |
Cys6 |
Tyr200 (el2) |
VDW |
|
28 |
Pro7 |
Thr202 (el2) |
VDW |
|
29 |
Pro7 |
Tyr200 (el2) |
VDW |
|
30 |
Leu8 |
Ile201(el2) |
VDW |
|
31 |
Leu8 |
Ile204 (helix 5) |
VDW |
|
32 |
Leu8 |
Trp203 (el2) |
VDW |
|
33 |
Leu8 |
Thr202 (el2) |
VDW |
|
34 |
Gly9 |
Gln295 (helix 6) |
VDW |
|
35 |
Gly9 |
Ser298 (el3) |
VDW |
|
36 |
Gly9 |
Trp297 (el3) |
HB |
|
37 |
Gly9 |
Val294 (helix 6) |
HB |
a HB, hydrogen bonding interaction.
b VDW, van der Waals attractive interaction.
Molecular modeling of other groups in combination with mutagenesis investigations reveals a large number of potential binding sites in OTR that are essential for ligand binding [1]. The comparison with the binding sites in our receptor model shows the following coincidences: Val115, Gln119, Met123, Gln171, Thr202, Val294, Gln295, Trp300, Glu307, Ile313, Val314, four of them - Gln119, Gln171, Gln295 and Glu301 – are in agreement with the experimental data as well.
3.5. Agonist binding induced conformational changes in the OTR
The receptor molecule can exist in two states (inactive and active), that are in conformational equilibrium. The intracellular portion of the receptor participate in interactions with the G protein. The conformational equilibrium is shifted to active receptor state after binding of the ligand in transmembrane pocket near to extracellular part. Binding of the agonist (ligand) to the receptor results in conformational changes in its extracellular and intracellular loops, as well as in its transmembrane portion [45 - 48]. A common mechanism of activation, in which participate highly conservative amino acid residues, is characteristic of the GPCRs members. These amino acid residues are located in the intracellular parts of helices TM3 (Arg137, which is part of the E(D)RY motif) and TM6 and form "ionic lock", which hold TM3 and TM6 in their inactive form. The disruption of intermolecular interactions between these residues is a key moment in activation process of the receptor. The observed conformational changes are: screening of the electrostatic interaction between the helices, motion of helices TM6, TM5 and divided of cytoplasmic parts of TM3 and TM6 after breakage of the "ionic lock" between them [49]. According to the polar pockets hypothesis the arginine of the motif E(D)RY is constrained in a pocket, in the inactive receptor conformation.
Conserved polar residues located on TM1, TM2 and TM7 form this pocket. In contrast, in the active receptor state triggered by the agonist, the arginine side chain is shifted outside the polar pocket. The negatively charged aspartic acid in the E(D)RY motif is also important, because from its two states, protonated-(neutral) and deprotonated- (anionic), depends the equilibrium between active and inactive receptor forms. These observations are confirmed by the results of mutagenesis studies and as well as by molecular dynamic simulations [50].
In order to investigate the GPCR activation mechanism two MD simulations were carried out for an unbound OTR (300 ns) and for an agonist (oxytocin) bound receptor (100 ns). From the oxytocin bound trajectory its follows that transmembrane helices TM1, TM2, TM6 (after slight initial rotation in the process of oxytocin binding) and TM7 remain stable (Figure 9). Unfolding of the helical structure of the TM3 (Arg113 – Met123) triggered by oxytocin binding at positions Val115, Glu119 and Met 123 is observed. The helical structures in TM4 (Val166-Leu177) and TM5 (Trp203- Leu206) are affected significantly in result of hormone interactions with EL2 (Tyr200, Ile201, Thr202 and Trp203), TM4 (Gln171) and TM5 (Ile204).
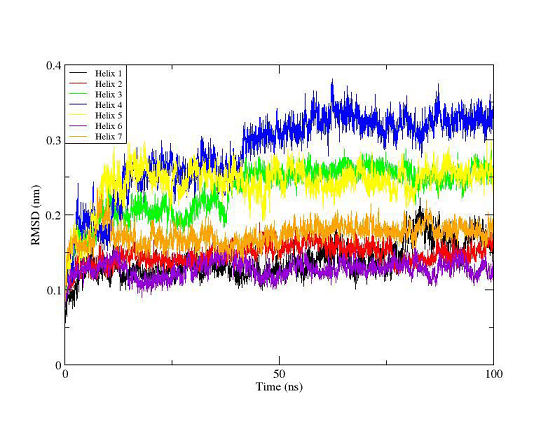
Figure 9. RMSD plot of the 7 transmembrane helices for the trajectory of the agonist- OTR complex. The colors indicate helix 1 (black), helix 2 (red), helix 3 (green), helix 4 (blue), helix 5 (yellow), helix 6 (violet) and helix 7 (orange).
The unbound OTR is characterized by a persistent intra-helix charge-reinforced H-bond between Arg137 (helix 3) of the E/DRY motif and the adjacent aspartate, Asp136. The structural changes in helices TM3 and TM4 result in breakage of this H- bond (Figure 10). At the same time the helical structure of the E/DRY motif is stabilized (Figure 11) and is shifted outside the polar pocket formed by the stable helices TM1, TM2 and TM7 (Figure 12). We do not observe any other changes in the receptor structure in the intracellular region.
Based on these observations we come to hypothesis that the shift of the E/DRY motif influences the G-protein and starts the process of its disintegration from the receptor and the corresponding intracellular process.
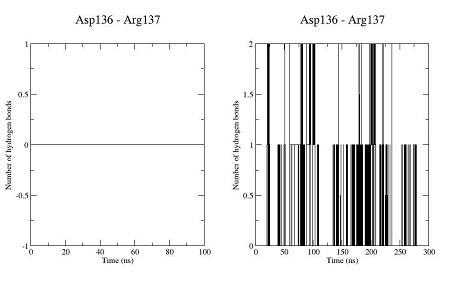
Figure 10. Number of hydrogen bonds between residues Asp136 and Arg137 for the trajectory of the unbound OTR (left) and the trajectory of the agonist-OTR complex (right).
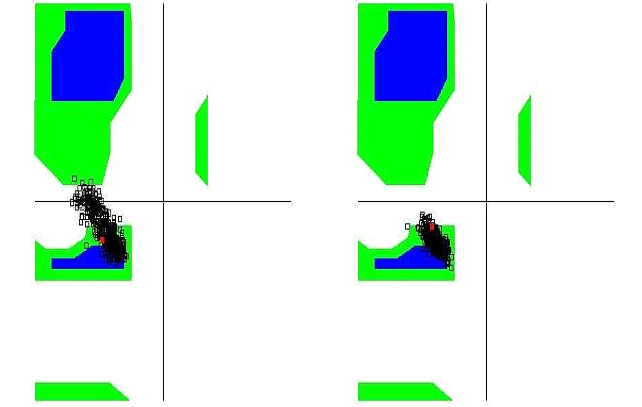
Figure 11. Ramachandran diagram of the Arg137 of the unbound OTR (left) and of the agonist-OTR complex (right).
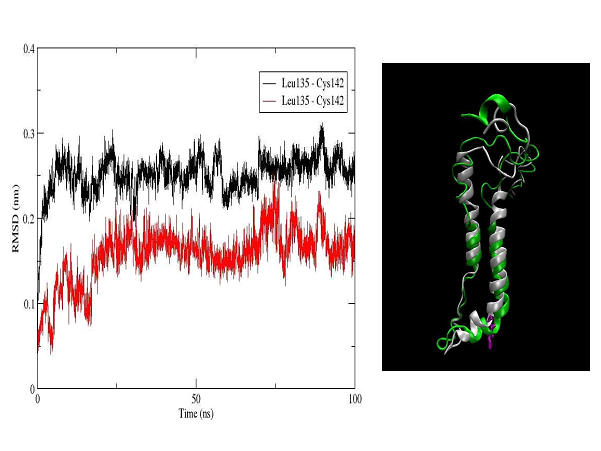
Figure 12. RMSD plot of the amino acid sequence Leu135-Cys142, which includes the E/DRY motif (Leu135-Asp136-Arg137-Cys138) in the OTR (left). The colors indicate the unbound OTR (black) and the agonist-OTR complex (red). In right panel is shown a superimposed structure of the helix 3 (right) and 4 (left) for the unbound OTR (white) and the agonist-OTR complex (green) - RMSD 3.5 Å. In violet is shown Arg137, which is part of transmembrane helix 3 and E/DRY motif.
In cases, where there are no crystal structures available of the target proteins, the homology models of GPCRs are highly valuable in drug-designing process [51]. In our study a 3D model of the human oxytocin G-protein coupled receptor was built based on homology modeling. The obtained 3D structure exhibited minor deviations (Cα RMSD = 1.05 Å) from the experimentally determined 3D structure of bovine rhodopsin that was used as a template. The additional MD energy minimization of the receptor did not lead to significant deviation from the initial structure. The receptor was embedded in a new improved model of the myometrial membrane (containing five different types of phospholipids). This biomolecular complex was used for investigation of the mechanism of oxytocin binding to its receptor.
A new rigid structure, a complex between oxytocin and a metal ion was built in order to take into account the influence of the Zn2+ ions on the hormone binding process. Stability of the complex was confirmed by series of MD simulations. The metal ion changes the 3D structure of the oxytocin and makes possible its binding with the receptor [34].
The complex of oxytocin bound to its receptor embedded in the uterine membrane was used to define the agonist receptor interaction sites and to investigate the mechanism of receptor activation. We found 21 potential binding sites, out of them 11 (essential for the binding) coincide with results of similar investigations by other groups and four of them Gln119, Gln171, Gln295 and Glu301 are in agreement with the experimental data as well. In nine cases was observed formation of H-bonds. The other sites are due to van der Waals interactions between amino acid residues of oxytocin molecule and the receptor.
The investigation of the structural changes in the receptor induced by the oxytocin binding shoed that the transmembrane helices TM1, TM2, TM6 and TM7 are stable when the helices TM3, TM4 and TM5 are partially unfold. The oxytocin – receptor interaction stabilizes the helical structure of the E/DRY motif and shifts it outside the polar pocket formed by the stable helices TM1, TM2 and TM7. This result supports the hypothesis for the critical role of the E/DRY motif in the receptor activation and is consistent with the experimental results for the A GPCRs family [47, 49].
Thus, the validated oxytocin receptor model is suitable for dynamical investigations of the ligand-receptor binding process and therefore, for the design of new highly selective antagonists with possible application for prevention of the premature births.
MD simulations were performed at the parallel cluster of the Atomic Physics Department at Faculty of Physics of Sofia University “St. Kl. Ohridski”. This work was supported in part by the CEEPUS programme and Austrian Agency for International Cooperation in Education and Research (OeAD - GmbH).Thanks to funding by this programme, some of the studies were conducted at the Center for Medical Statistics, Informatics and Intelligent Systems, Medical University of Vienna, Austria. We acknowledge the support from the Bulgarian National Science Fund (Grant DNTS-Austria-01/2/2013). We would like to thank Prof. Wolfgang Schreiner and Dr. Michael Kenn for their help. The authors are also grateful to Assoc. Prof. Rudolf Karch and Prof. Harald Heinzl for the carefully reading of the manuscript and for the helpful discussions.
Gimpl, G., Fahrenholz, F. The oxytocin receptor system: structure, function, and regulation. Physiol. Rev. 81 (2001) 629-683. PMid:11274341
View Article PubMed/NCBIFredriksson, R., Lagerström, M., Lundin, L., Schiöth., H. The G protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63 (2003) 1256-1272. PMid:12761335
View Article PubMed/NCBIGloriam, D., Fredriksson, R., Schiöth, H. The G protein-coupled receptor subset of the rat genome. BMC Genomics 8 (2007) 338. PMid:17892602
View Article PubMed/NCBIVrachnis, N., Malamas, F., Sifakis, S., Deligeoroglou, E., Iliodromiti, Z. The Oxytocin - Oxytocin Receptor System and Its Antagonists as Tocolytic Agents. Int. J.Endocrinol. 2011/ (2011) 1-8. PMid:22190926
View Article PubMed/NCBISanborn, B. M, Ku, C. Y., Shlykov, S., Babich, L. Molecular signaling through G- protein-coupled receptors and the control of intracellular calcium in myometrium. J. Soc. Gynecol. Investig. 12 (2005) 479-487. PMid:16202924
View Article PubMed/NCBILagerström, M. C., Schiöth, H. B. Structural diversity of G protein-coupled receptor and significance for drug discovery. Nat. Rev. Drug. Discov. 7 (2008) 339-357. PMid:18382464
View Article PubMed/NCBIOverington, J.P., Al-Lazikani, B., Hopkins, A. L. How many drug targets are there? Nat. Rev. Drug. Discov. 5 (2006) 993-996. PMid:17139284
View Article PubMed/NCBISvoboda, P., Teisinger, J., Novotny, J., Bourova, L., Drmota, T., Hejnova, L., Moravcova, Z., Lisy, V., Rudajev, V., Stöhr, J., Vokurkova, A., Svandova, I., Durchankova, D. Biochemistry of transmembrane signalling mediated by trimeric G proteins. Physiol. Res. 53 (2004) S141-S152.
Lomize, A. L., Pogozheva, I. D., Mosberg, H. I. Structural organization of G - protein coupled receptors. J. Comput. Aided. Mol. Des. 13 (1999) 325-353. PMid:10425600
View Article PubMed/NCBIBöhm, Hans-Joachim, Klebe, G., Kubinyi, H. Wirkstoffdesign Der Weg zum Arzneimittel, 1st edn. Spektrum Akademischer Verlag, Heidelberg. ISBN 13 (1996) 978-3827413536
Krieger, E., Nabuurs, S., Vriend, G. Homology modeling. Structural Bioinformatics 25 (2003) 507-521. PMid:12647402
View Article PubMed/NCBIWieman, H., Tøndel, K., Anderssen, E., Drabløs, F. Homology - Based Modelling of Targets for Rational Drug Design. Mini Rev. Med. Chem. 4 (2004) 793-804.
View ArticleHalip, L., Cruia, A., Borota, A., Maria Mracec, Curpan, R. F., Mircea Mracec. 3D Homology model of the α2c-adrenergig receptor subtype. Revue Roumaine de Chimie 57 (2012) 763-768.
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic. Acids. Res. 43(Database issue) (2015) D6-17. PMid:25398906
View Article PubMed/NCBIAltschul, S.F, Gish, W., Miller, W., Myers, E. W., Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215 (1990) 403-410. 80360-2
View ArticleRose, P., Prlic, A., Altunkaya, A., Bi, C., Bradley, A., Christie, C. H., Costanzo, L. D., Duarte, J. M., Dutta, S., Feng, Z., Green, R. K., Goodsell, D. S., Hudson, B., Karlo, T., Lowe, R., Peisach, E., Randle, C., Rose, A. S., Shao, C., Tao, Y. P., Valasatava, Y., Voigt, M., Westbrook, J. D., Woo, J., Yang, H., Young, J. Y., Zardecki, C., Berman, H. M., Burley, S. K. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic. Acids. Res. 45 (Database issue) (2017) D271-D281.
View ArticleYeagle, P., Choi, G., Albert, A. Studies on the structure of the G-protein-coupled receptor rhodopsin including the putative G-protein binding site in unactivated and activated forms. Biochemistry 40 (2001) 11932 - 11937. PMid:11570894
View Article PubMed/NCBISali, A., Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology 234 (1993) 779-815. PMid:8254673
View Article PubMed/NCBIShen, M. Y., Sali, A. Statistical potential for assessment and prediction of protein structures. Protein. Sci. 15 (2006) 2507-2524. PMid:17075131
View Article PubMed/NCBIJo, S., Kim, T., Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2 (2007) e880. PMid:17849009
View Article PubMed/NCBIJo, S., Lim, B., Klauda, J. B., Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J . 97 (2009) 50-58. PMid:19580743
View Article PubMed/NCBIWoolf, T. B., Roux, B. Structure, energetics, and dynamics of lipid-protein interactions: a molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins: Structure., Function, and Genetics 24 (1996) 92-114. 1097-0134(199601)24:1<92::AID-PROT7>3.0.CO;2-Q
View ArticleAbracham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelizm from laptops to supercomputers. Software X 1-2 (2015) 19-25.
View ArticleHuang, J., MacKerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34 (2013) 2135-2145. PMid:23832629
View Article PubMed/NCBISteinbach, P. J., Brooks, B. R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15 (1994) 667-683.
View ArticleEssmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., Pedersen, L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 103 (1995) 8577-8593.
View ArticleMadhusudhan, M. S., Marti-Renom, M. A., Sanchez, R., Sali, A. Variable gap penalty for protein sequence-structure alignment. Protein Eng. Des. Sel. 19 (2006) 129-133. PMid:16423846
View Article PubMed/NCBISchlegel, B., Sippl, W., Höltje, H-D. Molecular dynamics simulations of bovine rhodopsin: influence of protonation states and different membrane-mimicking environments. J. Mol. Model. 12 (2005) 49-64. PMid:16247601
View Article PubMed/NCBICarroll, P. M., Sereda, D. D. Cell Membrane of Uterine Smooth Muscle. Nature 217 (1968) 666-667. PMid:4866539
View Article PubMed/NCBIPearlmutter, A. F., Soloff, M. S. Characterization of the Metal Ion Requirement for Oxytocin-Receptor Interaction in Rat Mammary Gland Membranes. J. Biol. Chem. 254 (1979) 3899-3906.
Kozlowski, H., Radomska, B., Kupryszewski, G., Lammek, B., Livera, C., Pettit, L. D., Pyburn, S. The unusual co-ordination ability of vasopressin-like peptides; potentiometric and spectroscopic studies of some copper(II) and nickel(II) complexes. Journal of the Chemical Society, Dalton Transactions 1 (1989) 173-177.
View ArticleBal, W., Kozlowski, H., Lammek, B., Pettit, L. D., Rolka, K. Potentiometric and spectroscopic studies of the Cu(II) complexes of Ala-Arg8-vasopressin and oxytocin: two vasopressin-like peptides. J. Inorg. Biochem. 45 (1992) 193-202. 80044-V
View ArticleDanyi, P., Varnagy, K., Sovago, I., Schon, I., Sanna, D., Micera, G. Potentiometric and spectroscopic studies of the copper (II) complexes of peptide hormones containing disulfide bridges. J. Inorg. Biochem. 60 (1995) 69-78. 00003-7
View ArticleMiladinova, E. Molecular dynamic study of the stability of oxytocin - divalent zinc complex in aqueous solution. Journal of Computational Chemistry and Molecular Modelling 3 (2019) 252-260.
View ArticleLiu, D., Seuthe, A., Ehrler, O., Zhang, X., Wyttenbach, T., Hsu, J., Bowers, M. Oxytocin - Receptor Binding: Why Divalent Metals Are Essential. Journal of the American Chemical Society 127 (2005) 2024-2025. PMid:15713062
View Article PubMed/NCBIHumphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics 14 (1996) 33−38. 00018-5
View ArticleCerqueira, N. M. F. S. A., Ribeiro, J., Fernandes, P. A., Ramos, M. J. vsLab-An implementation for virtual high-throughput screening using AutoDock and VMD. International Journal of Quantum Chemistry 111 (2010) 1208−1212.
View ArticleKumari, R., Kumar, R., Lynn, A. g_mmpbsa - A Gromacs tool for high- throughput MM-PBSA calculations. J. Chem. Inf. Model. 54 (2014) 1951−1962. PMid:24850022
View Article PubMed/NCBIDurrant, J., McCammon, J. A. HBonanza: A computer algorithm for molecular- dynamics-trajectory hydrogen - bond analysis. J. Mol. Graph. Model. 31 (2011) 5-9. PMid:21880522
View Article PubMed/NCBISluzars, M. J., Sluzars, R., Ciarkowski, J. Molecular dynamics simulaton of human neurohypophyseal hormone receptors complexed with oxytocin - modeling of an activated state. J. Pept. Sci. 12 (2006) 171 -179. PMid:16114099
View Article PubMed/NCBIPostina, R., Kojro, E., Fahrenholz, F. Separate agonist and peptide antagonist binding sites of the oxytocin receptor defined by their transfer into the V2 vasopressin receptor. J. Biol. Chem. 271 (1996) 31593-31601. PMid:8940177
View Article PubMed/NCBIMouillac, B., Chini, B., Balestre, M., Elands, J., Trumpp-Kallmeyer, S., Hoflack, J., Hibert, M., Jard, S., Barberis, C. The binding site of neuropeptide vasopressin in V1a receptor. Evidence for a major localization within transmembrane regions. J. Biol.Chem. 270 (1995) 25771-25777. PMid:7592759
View Article PubMed/NCBIFanelli, F., Barbier, P., Zanchetta, D., De Benedetti, P., Chini, B. Activation mechanism of human oxytocin receptor: a combined study of experimental and computer simulated mutagenesis. Mol. Pharmacol. 56 (1999) 214-225. PMid:10385703
View Article PubMed/NCBIKoehbach, J., Stockner, T., Bergmayr, C., Muttenthaler, M., Gruber, C. Insights into the molecular evolution of oxytocin receptor ligand binding. Biochem. Soc. Trans. 41 (2013) 197-204. PMid:23356283
View Article PubMed/NCBIStrader, C. D., Fong, T. M., Tota, M. R., Underwood, D., Dixon, R. A. Structure and function of G protein-coupled receptors. Annu Rev. Biochem. 63 (1994) 101-132. PMid:7979235
View Article PubMed/NCBIWeiss, J. M., Morgan, P. H., Lutz, M. W., Kenakin, T. P. The cubic ternary complex receptor-occupancy model III resurrecting efficacy. J. Theor. Biol. 181 (1996) 381-397. PMid:8949584
View Article PubMed/NCBIRubenstein, L. A., Lanzara, R. G. Activation of G protein-coupled receptors entails cysteine modulation of agonist binding. Journal of Molecular Structure: Theochem 430 (1998) 57-71. 90217-2
View ArticleMirzadegan, T., Benko, G., Filipek, S., Palczewski, K. Sequence analyses of G protein-coupled receptors: similarities to rhodopsin. Biochemistry 42 (2003) 2759- 2767. PMid:12627940
View Article PubMed/NCBIBallesteros, J. A., Jensen, A. D., Liapakis, G., Rasmussen, S. G., Shi, L., Gether, U., Javitch, J. A. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol.Chem. 276 (2001) 29171-29177. PMid:11375997
View Article PubMed/NCBIChini, B., Fanelli, F. Molecular basis of ligand binding and receptor activation in the oxytocin and vasopressin receptor family. Experimental Physiology 85 (2000) 59S- 66S. PMid:10795907
View Article PubMed/NCBIKoehl, P., Levitt, M. A brighter future for protein structure prediction. Nat. Struct. Biol. 6 (1999) 108-111. PMid:10048917
View Article PubMed/NCBI